

“The last three generations of my family were struck with Parkinson’s disease”
The Westmeath United Church records state that my great grandfather died of “paralysis” at the age of 63, over 100 years ago. My grandfather, father and uncle were all later diagnosed with Parkinson’s disease, which led my family to develop a long-standing and intimate relationship with the disease.
Throughout my journey as a caregiver to my dad I was toggling between the hope of new discovery as I read about, and contributed to, exciting developments on the research side, and the sad truth that the therapies we sought were not enough. We just didn’t know enough about the underlying mechanisms of this disease. The main therapy had been discovered decades ago, and the newer technologies like deep brain stimulation were highly restricted in what kinds of patients were eligible. My dad was a farmer and a mechanic and he desperately wanted someone to open up his brain and fix it. But as he always said to me “you can’t fix what you don’t understand”.
In my childhood he would say this about engines and combines, but he returned to this refrain after his diagnosis. I was proud when he attended my session at the 3rd World Parkinson Congress in Montreal back in 2013. At least I could show him that I was trying to understand, that we were working together as a field to offer him hope and future cures.
My entry into Parkinson’s research coincided with a series of studies in the mid-2000s showing that at least two specific PD-related proteins PINK1 and Parkin were linked to mitochondrial dynamics and quality control in fly models of disease. This prompted a team of Parkinson’s scientists in Montreal and Ottawa to ask me to join them in a collaborative grant from Brain Canada to explore the mitochondrial contribution to PD.
As a mitochondrial cell biologist my team had discovered that mitochondria could shed vesicles, and we knew these vesicles could target damaged mitochondrial proteins to the late endosome/lysosome. Dr. Ted Fon at The Neuro (Montreal Neurological Institute-Hospital) had shown earlier that Parkin could regulate vesicle transport from the cell surface, so we began to work together to test whether PINK1 and Parkin might regulate mitochondrial vesicle transport.
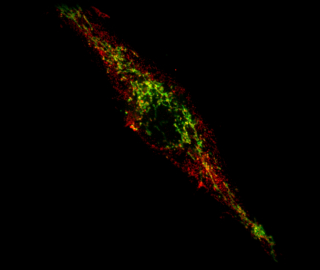 While the field exploded in 2008 with Richard Youle’s discovery that PINK1 and Parkin can mediate mitophagy (the digestion of entire mitochondrial organelles), we continued our work into the mechanisms and function of more targeted mitochondrial protein degradation. With Ted we eventually published a number of papers outlining this pathway and the contribution of PINK and Parkin to the removal of selective mitochondrial proteins for degradation.
While the field exploded in 2008 with Richard Youle’s discovery that PINK1 and Parkin can mediate mitophagy (the digestion of entire mitochondrial organelles), we continued our work into the mechanisms and function of more targeted mitochondrial protein degradation. With Ted we eventually published a number of papers outlining this pathway and the contribution of PINK and Parkin to the removal of selective mitochondrial proteins for degradation.
In the meantime the field of Parkinson’s research was pushing into more complex models. It was becoming clear that the immune system has a part to play in the etiology of disease progression, with evidence of innate, adaptive and microbiome contributions. We were also realizing in my own lab that mitochondrial vesicles are very plastic, and that the triggers that drive them, the contents within them, and the fate that awaits them is highly selective and tightly regulated. As we were trying to understand these complexities, an old immunologist friend of mine Michel Desjardins asked me to advise him on his project investigating how mitochondrial antigens are presented within the thymus to establish immunotolerance.
After a few years of exciting experimentation done primarily by Michel’s postdoc Diane Matheoud and Ayumu Sugiura in my lab, we learned that mitochondrial vesicles were activated by heat stress or infection with bacteria, a process that was critical for mitochondrial antigen presentation. Linking this to Parkinson’s disease, the PD related proteins PINK1 and Parkin actively repressed this pathway. This suggested that PINK1 and Parkin had broader functions in disease than mitochondrial quality control. It led us to hypothesize that patients carrying mutations in these proteins may have an increased presentation of mitochondrial antigens upon infection or stress, which could drive an autoimmune reaction ultimately linking to the destruction of dopaminergic neurons. But how could we test this?
We needed collaborators who understood immunology, infection and dopaminergic neurons. So we expanded our team and began to infect wild type and PINK1 knock-out mice with gut E.coli called Citrobacter rodentium. This is a relatively mild infection that resolves relatively quickly, just like when we get infected from contaminated food. We knew from cultured macrophage work that this bacterium could drive high levels of mitochondrial antigen presentation; it was also the primary model system used by an old friend from graduate school, Samantha Gruenheid.
Unlike patients lacking PINK1 who have early onset disease, PINK1 knockout mice show almost no phenotype. The infection process itself seemed similar in both types of mice, meaning they cleared the infection and recovered similarly. But we could see that primary macrophages and dendritic cells from the PINK1 knock-out mice presented high levels of mitochondrial antigens, and the T-cell repertoire in the mice was changing to specifically target mitochondrial antigens. Ultimately Tyler Cannon, a brilliant young master’s student in Samantha’s lab leading the project with Diana Matheoud, came to us with videos of the previously infected PINK1 KO mice. He said “I think something is wrong with these mice. They barely move!” He was showing us mice that had cleared the infection about 2 months earlier, so we were immediately excited! However, they could be “frozen” for lots of reasons.
We approached Louis-Eric Trudeau to help resolve this question, an electrophysiologist and neuroscientist who’s worked in PD for decades. We treated these mice with L-Dopa and they recovered their “frozen” phenotype within 30 minutes, just like with PD patients. A close analysis of the dopaminergic neurons revealed a loss of the neuron terminals, but the cell bodies remained intact. We were again surprised after another 6 months passed and the mice fully recovered, including the return of the dopaminergic terminal projections into the striatum. This meant that we had developed a proof-of-concept study in a genetically susceptible mouse model where infection leads to a transient Parkinsonism. While mice are often derided as terrible models of human disease we had made an important first step. It was consistent with others who were exploring the roles of immunity in Parkinson’s, and gave us a new framework to investigate. We published our findings with all four of us as corresponding authors, highlighting the importance of our multidisciplinary team.
In the end we had gone from a basic cell biological observation of the mechanisms of antigen presentation in macrophages to a global mouse model. However, this opens a pandoras box of questions. What is the order of events along the gut-brain axis in this system? What cell types and tissues are the PINK1 and Parkin proteins most important for and how do they really regulate the immune system? Is the microbiome involved? What about the blood-brain barrier issue? These in addition to a host of basic mechanistic questions about immune signaling, the mitochondrial response, and the overarching question of whether and how PINK1 action is linked to quality control or signaling? We also wondered whether other PD related proteins like LRRK2, VPS35, synuclein, GBA act in a similar pathway, or are our insights unique to PINK1 and Parkin? And herein lies the major challenge in the field at the moment. No one lab can handle this level of complexity. I agree whole heartedly with Malu Tansey who famously said “Science is a team sport”.
Our team has now expanded to nine core members as we continue our work to unravel the complex strands of this disease. We are thrilled to work with incredibly talented young investigators with skill sets I could only dream of. Jo Anne Stratton and Janelle Drouin-Ouellet are experts in single cell sequencing and stem cell research, and the team is expanding to test whether some of the concepts we are exploring may hold true in patient populations. We were extremely fortunate to have been awarded a new team grant from Aligning Science Across Parkinson’s (ASAP), which opens new avenues of collaboration and open discussions all over the world.
It is always a challenge to maintain and nurture meaningful collaborations. First there is a language and education issue that takes a while to overcome. As a basic cell biologist it is daunting to learn the details of the immune system, the gut response to infection, stem cell reprogramming, and the many unique features of the dopaminergic neuron. I am grateful for the patience of my collaborators! The second major challenge is a broader social issue in the scientific enterprise. Funding and promotion mechanisms tend to value the first and last author positions almost exclusively, discounting collaborative studies as demonstrating a “lack of independence”. This doesn’t bother me personally since I’m a long way into my career, but for the early career investigator it can be deadly.
I do see a slow sea-change in these views, which is hopeful, and I am grateful for programs like ASAP that actively push against the singular narrative of the lone genius scientist and instead promote open science and collaboration. The Neuro, my own research institution at McGill University, has also been leading the way into open science platforms and policies to drive research faster and push for therapies and cures to many neurological diseases.
The last three generations of my family were struck with Parkinson’s disease. The treatments barely changed between my grandfather’s and father’s time, which tells me that scientists were looking down the wrong rabbit hole. The field has exploded into areas beyond neuroscience into a more holistic view of the long, degenerative process of PD. I am hopeful we may soon be in a position to answer my father’s question and tell him that we DO understand, and we CAN fix it!

This blog was originally published by the World Parkinson’s Congress.