
Combining molecules may prove to be key

A new study conducted in the lab of Dr. Gergely Lukacs at McGill University’s Faculty of Medicine may offer a more effective avenue to explore the treatment of cystic fibrosis. The findings are published in Nature Medicine.
Cystic fibrosis (CF) mutations impair the function of the cystic fibrosis transmembrane conductance regulator (CFTR), a membrane protein encoded by the CF gene, which ensures proper salt and water content of fluids lining the gut and lung inner surfaces. CF mutations lead to lung infection and uncontrolled inflammation, the primary cause of premature death in CF patients.
The most common of these mutations, single amino acid deletion of phenylalanine at position 508 (ΔF508) in one of the five domains of CFTR, causes serious damage in the three dimensional architecture of the CFTR protein and, as a consequence, increases the stickiness of the surface liquid layer on epithelial cells that protect body tubes (e.g. airway and gut) from environmental insults (e.g. bacteria and virus particles) and the secretory portions of glands and their ducts.
While currently available drugs only modestly correct mutation-caused CFTR folding defects, in their study, the researchers showed that by selecting a combination of small molecules to target multiple, distinct structural defects they can potently rescue the mutant CFTR architecture and function in airway epithelia. “While individually these compounds marginally improve ΔF508-CFTR folding efficiency, plasma membrane function, and stability, combining them leads to approximately 50-100% of wild type correction in cell lines and primary cells isolated from the nose of CF patients, as well as in a mouse model of cystic fibrosis,” explains Dr. Guido Veit, a Research Associate in Dr. Lukacs’ lab and the study’s lead author. Remarkably, the small molecule cocktails also rescue the functional defects of some rare CF mutations resistant to available drugs.
The work builds on a 2012 study published in Cell by Dr. Lukacs’ lab that identified that ΔF508 mutation leads to at least two distinct structural defects in CFTR and that both need to be corrected. Since that time they had been trying to identify small molecule compounds that could serve as correctors with distinct mechanism of action. To achieve this goal they screened for compounds in the background of one corrector with a known mechanism of action in order to identify correctors with complementary mechanisms. The screening of 600,000 small molecules was performed in collaboration with the Genomics Institute of the Novartis Research Foundation (GNF). Subsequent mechanistic work led to the identification of a corrector combination with twice the efficacy of the current FDA-approved corrector therapy in cells isolated from homozygous ΔF508 patients.
The compounds have not yet been optimized for their pharmacological properties, a prerequisite for clinical trials but the researchers are working on this and are planning to connect with partners in order to develop the compounds into corrector drugs. “Our hope is that by increasing the correction efficacy of ΔF508-CFTR to about 50% of the wild type level, the clinical manifestations of cystic fibrosis in individuals that are homozygous for ΔF508, which comprises approximately 50% of all CF patients, can be completely alleviated through this therapy since heterozygous carriers lack disease symptoms,” explains Dr. Lukacs, who is a Professor in the Departments of Physiology and Biochemistry and Canada Research Chair in Cystic Fibrosis and Conformational Diseases at McGill. “Additionally, because of the similarity between the CFTR architecture and other members of the ABC transporter protein family, therapeutic approaches for other diseases such as neonatal respiratory distress syndrome, familial hyperinsulinism, and progressive familial intrahepatic cholestasis may also benefit from our findings down the line.”
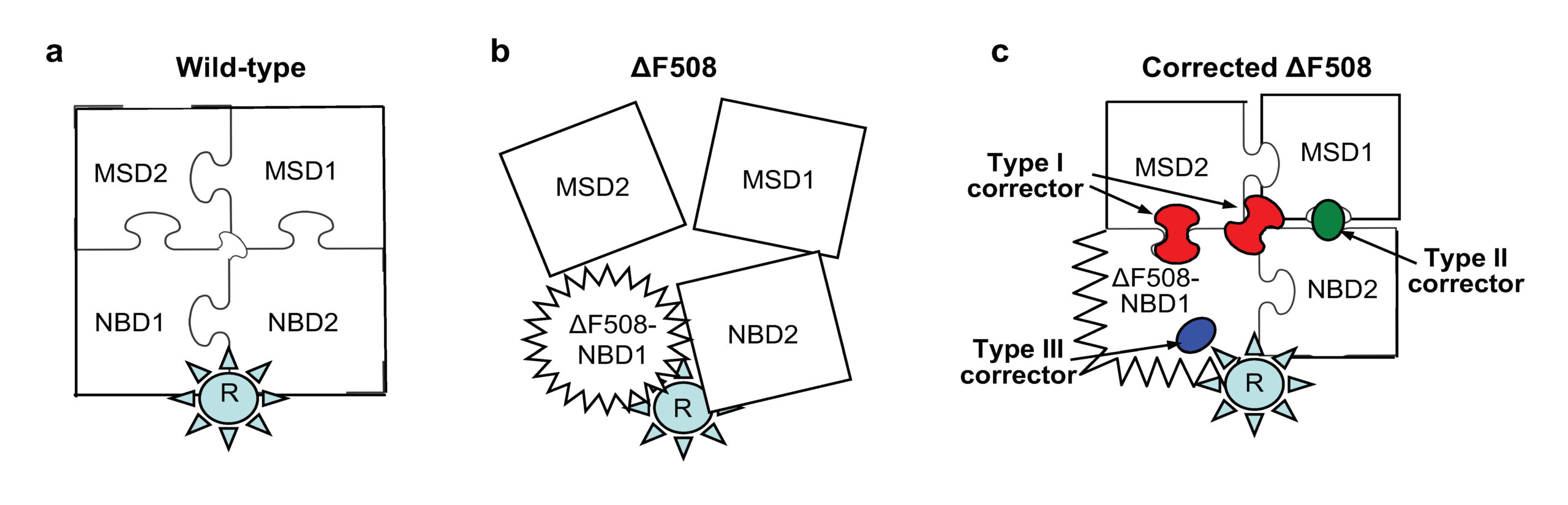
Importantly, this strategy also worked for other point mutations in various domains that cause CFTR misfolding. This underscores the interdependent nature of CFTR domain folding/assembly (i.e. the puzzle pieces only adopt their final shape when they fit together) and the allosteric or long range conformational coupling of corrector molecules (i.e. a given corrector promotes folding of one domain, which in turn facilitates the spatial fitting of other pieces of the puzzle).
This work was performed in collaboration with Drs. Elie Matouk and Saul Frenkiel, McGill University Health Centre, Department of Respiratory Medicine and Department of Otolaryngology-Head and Neck Surgery, McGill, respectively, Drs. I. Sermet-Gaudelus, A Edelman and E Dreano, Necker Institute, Paris, and Dr. W Barnes group, GNF, San Diego.
The research was supported by Cystic Fibrosis Canada, Cystic Fibrosis Foundation Therpeautic Inc., CIHR and NIH.
“Structure-guided combination therapy to potently improve the function of mutant CFTRs” by Guido Veit et al. and Gergely Lukacs, is published in Nature Medicine.
DOI: https://dx.doi.org/10.1038/s41591-018-0200-x
October 11, 2018